Átfogó elemzésben vizsgálták az ELTE Komplex Rendszerek Fizikája Tanszék munkatársai, mennyire gyakori, hogy valaki több különböző SARS-CoV-2 variánssal fertőződik meg egyidejűleg. A vizsgálat során kitértek arra is, hogy ezekben az esetekben milyen eséllyel jöhet létre a különböző variánsokból egy új, hibrid vírusváltozat.
A COVID-19 betegséget okozó SARS-CoV-2 koronavírusról a világon korábban nem látott mértékű monitorozásnak és nemzetközi összefogásnak köszönhetően óriási adatmennyiség gyűlt össze és érhető el az interneten, amely a folyamatos nyomon követésen felül számos alapkutatás kiindulásaként is szolgálhat. Csabai István kutatócsoportjának közreműködésével egy európai konzorcium létrehozta az Európai COVID-19 Adatportált, amelynek célja a nyilvánosan elérhető SARS-CoV-2 genomszekvenálási eredmények egységes módszertannal való elemzése és az eredmények könnyen kereshető adatbázisba építése. Világszinten több mint 2 millió mintát gyűjtöttek össze és dolgoztak fel a járvány kezdete óta.
Az adatbázis tartalmazza minden megszekvenált minta esetében a genomban előforduló mutációkat (módosulásokat) és azok előfordulási arányát is az adott mintát alkotó, tehát egy betegtől származó víruspopulációban. Ez azért is fontos lehet, mert a vírus evolúciója során az újonnan megjelenő mutációk gyakran először csak kis arányban vannak jelen (1a. ábra), de a következő generációkban elterjedhetnek, ha a mutáció előnyös a vírus számára. Az előfordulási arányokból (ún. allélfrekvencia értékekből) következtethetünk arra is, hogy egy mintában egyszerre több ismert variáns (pl. Alfa, Delta, Omikron stb.) is jelen van-e. Ha egy egyén egyszerre több variánssal is megfertőződik (koinfekció, 1b. ábra), az ugyan nem minden esetben, de okozhat súlyosabb tüneteket is, valamint a két variáns kombinációjából létrejöhet egy harmadik, ún. „rekombináns” vírusvariáns.
Az adatokat a kutatók Pipek Orsolya Anna és Medgyes-Horváth Anna vezetésével elemezték, az elemzést különlegessé teszi, hogy korábban csak néhány országban, néhány ezer minta bevonásával folytattak ilyen jellegű kutatást, világszinten és ilyen sok mintát együttesen azonban most először vizsgáltak.
A SARS-CoV-2 koronavírus variánsai mind eltérő genetikai állománnyal (szekvenciával) rendelkeznek, a kezdeti Wuhanban megjelent változathoz képest az elterjedt variánsok esetében több olyan egyedien jellemző (definiáló) mutáció fordul elő, ami más variánsokban nem. A kutatók ezeket a definiáló mutáció kombinációkat vizsgálták az Európai Covid-19 Adatportál szekvenciáiban, és keresték azokat a mintákat, amelyekben kis arányban is akár, de több variáns definiáló mutációi is előfordulnak.
A minták 0,35 százalékában (összesen 7700 esetben) találták meg egyszerre legalább két variáns jellemző mintázatait, a legtöbb esetben a Delta és az Omikron variánsok keveredtek egy mintán belül. Az egyidejű fertőződésekhez elengedhetetlen, hogy egyszerre több variáns is terjedjen a társadalomban, ez figyelhető meg a Delta és Omikron variánsok esetében 2021 vége és 2022 eleje között (1c. ábra). Ezek az koinfekciók kiemelkedően gyakoriak voltak Dél-Afrikában, ahol a HIV vírus okozta AIDS megbetegedések száma magas, ami megfelelő kezelés hiányában immunhiányos állapotot eredményez, és elhúzódó fertőzéseket okozhat. Egy ilyen elhúzódó fertőzés vezethetett például az Omikron variáns megjelenéséhez is: a hosszú fertőzés során az eddigi variánsokhoz képest jóval több mutáció következett be, létrehozva egy sokkal fertőzőképesebb változatot.
A hosszan elhúzódó esetek növelik a valószínűségét a koinfekciónak is, hiszen a hosszú ideig tartó lefolyásuk alatt könnyebben előfordulhat, hogy a beteg találkozik másik vírusvariánssal és elkapja azt. A legyengült immunrendszer ezen felül kedvez a vírus gyors szaporodásának is, ami növeli a rekombináció (genomi keveredés) valószínűségét, hiszen ez legnagyobb eséllyel úgy történik meg, hogy a vírus genetikai állományának másolása során (tehát szaporodás folyamán) véletlenül az egyik variáns genomjáról a másikra „ugrik”, létrehozva ezzel egy vegyes, hibrid genomot.
A kutatás célja volt az is, hogy megvizsgálják, végbement-e a rekombináció azokban a mintákban, amelyekben egyszerre több variáns is jelen van. Az ilyen jellegű vizsgálatokat azonban számos technikai nehézség hátráltatja. Először is a rekombináns verzió genomját (vagy annak részletét) a mintát alkotó víruspopuláció csak nagyon kis arányban tartalmazza, ami nagyban nehezíti a detektálását. További kihívást jelentett, hogy a genomszekvenálás során alkalmazott polimeráz-láncreakció (PCR) során véletlenszerűen keletkező, ún. „kiméra” szekvenciák jöhetnek létre, amelyek gyakorlatilag megkülönböztethetetlenek a valódi, a beteg szervezetében spontán megjelenő rekombináns szekvenciáktól. Nehézséget okozott az is, hogy a genom kevéssé mutált (variábilis) szakaszait érintő rekombinációs töréspontok gyakran kimutathatatlanok, mivel a különböző variánsok definiáló mutációi a genom mentén nem egyenletesen helyezkednek el (többségük például a vakcinagyártásban is használt tüskefehérjét kódoló részen).
A kutatók két eljárást fejlesztettek ki, amelyek ezeket a körülményeket figyelembe véve tudják a rekombinációt detektálni, és sikerült a Delta-Omikron koinfekciók esetében három olyan régiót is azonosítani, ahol gyakran történhet rekombináció. Az eredmény hozzájárulhat ahhoz, hogy az esetlegesen veszélyes, hibrid vírusokat még az elterjedésük előtt detektálhassák.
A kutatás az Európai Unió Horizon 2020 kutatási és innovációs program keretében a 874735 (VEO) és a 101046203 (BY-COVID) számú pályázatokból valósult meg, az elemzés a Nature Communications folyóiratban jelent meg.
Forrás: Eötvös Loránd Tudományegyetem, továbbította a Helló Sajtó! Üzleti Sajtószolgálat.Letölthető fotók
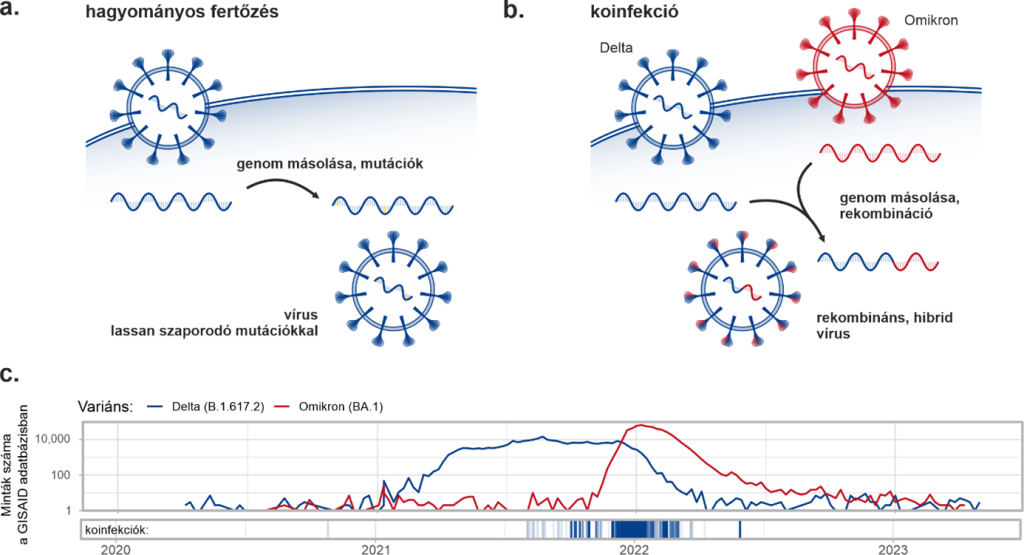
1a. Hagyományos megfertőződés esetén a vírus genetikai állományának másolása során szert tehet ugyan néhány mutációra, ezek eredményeként azonban egy nagyon hasonló, kevés módosulást tartalmazó vírusvariáns jön létre. 1b. Koinfekciókor a két variáns genomja másolás során összekeveredhet, rekombinálódhat, egy teljesen új, hibrid vírust hozva létre. 1c. A Delta és Omikron fertőzések számának alakulása (fent) a koinfekciókkal együtt ábrázolva (lent).


